Categoria: Bioinformática estrutural

Alinhamento de estruturas 3D com o software Multiprot
Reading Time: 3 minutes MultiProt é um software automatizado e eficiente para detecção de múltiplos alinhamentos estruturais de proteínas. Seu princípio básico visa encontrar os núcleos geométricos comuns entre as moléculas de entrada. Isso não exige que todas as moléculas de entrada participem do alinhamento, o que permite que o algoritmo seja mais rápido e…

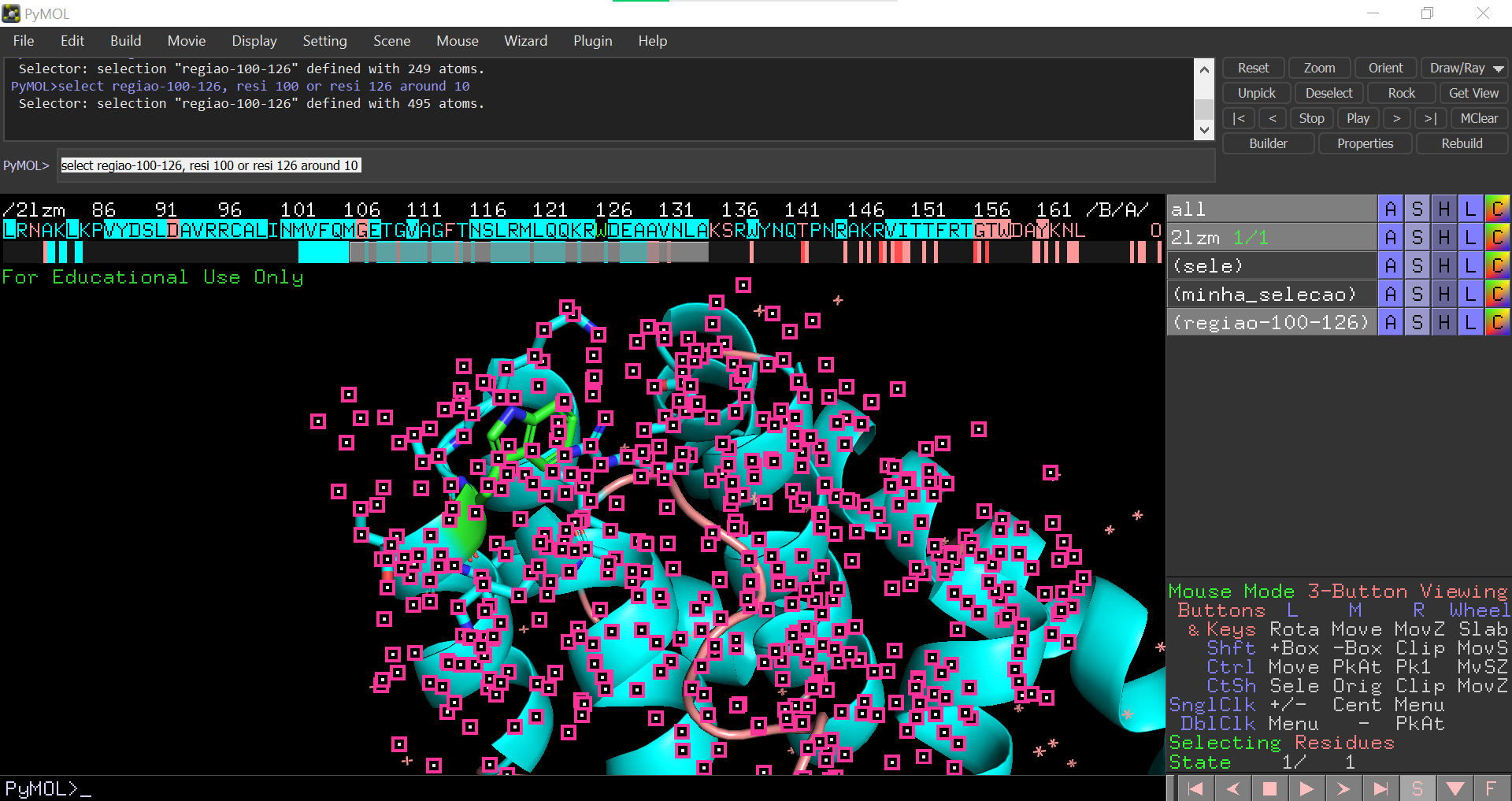
Selecionando partes de uma proteína com PyMOL (comando select)
Reading Time: 3 minutes O comando select pode ser usado no terminal de comandos do PyMOL para selecionar resíduos ou grupos de resíduos. Sintaxe: select [o que se deseja selecionar] Há várias palavras-chave que podem ser usadas na seleção, como por exemplo: resi: seleção com base no número do resíduo (e.g., select resi 20); resname:…
Tutorial: modelagem de proteínas usando MODELLER
Reading Time: 12 minutes Nesta seção será abordado a ferramenta MODELLER. O MODELLER é um software com vários pacotes criado por Andrej Sali e Tom L. Blundell em 1989 [14]. O MODELLER é uma ferramenta gratuita com uso restrito a linha de comando e não possui interface gráfica de usuário. Atualmente, o MODELLER utiliza a…
Modelagem de proteínas usando SWISS-MODEL
Reading Time: 4 minutes Nesta seção, será abordado a ferramenta SWISS-MODEL. Diferentemente do MODELLER, SWISS-MODEL é um servidor web que possui interface gráfica. Seu algoritmo também é diferente, pois utiliza regiões estruturais conservadas para construção dos modelos pelo método de união dos corpos rígidos. Porém, o SWISS-MODEL também parte do princípio de que proteínas homólogas…
Tutorial: modelagem de proteínas com I-TASSER
Reading Time: 6 minutes Um dos programas mais populares de Threading é o I-TASSER [28,30], que foi premiado diversas vezes na competição CASP (Critical Assessment of protein Structure Prediction). O I-TASSER (Figura 24) está disponível como um servidor web para predição automatizada de estrutura de proteínas e suas respectivas funções. A identificação dos moldes a…
Modelagem de proteínas ab initio com ROBETTA
Reading Time: 4 minutes A seguir, vamos utilizar o servidor web ROBETTA (http://robetta.bakerlab.org) para a modelagem de estruturas proteicas (Figura 33). O servidor utiliza a implementação automatizada do programa ROSETTA (https://www.rosettacommons.org/) no qual é possível realizar tanto modelagem comparativa quanto ab initio. A metodologia ROSETTA baseia-se em dividir a sequência em fragmentos de tamanho entre…

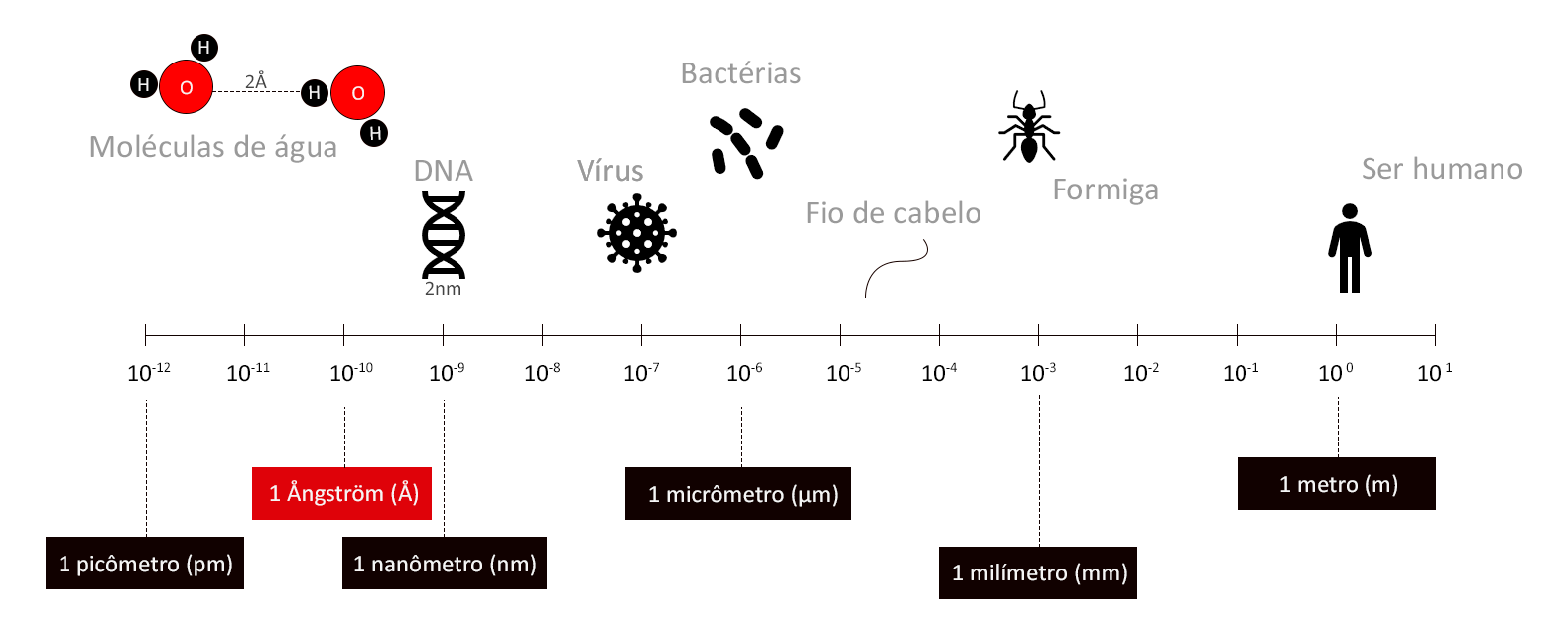
O que é ångström (Å) e quanto vale 1 Å?
Reading Time: < 1 minutes Ångström (Å) é uma unidade de medida equivalente a 10-10 m (ou seja, 1 Å = 0,0000000001 metros). Em geral, ångström é uma unidade utilizada para medidas que envolvem átomos. Curiosidades O nome é uma homenagem ao físico sueco Anders Jonas Ångström. O símbolo Å origina-se do alfabeto sueco e é composto…
Modelagem computacional de proteínas
Reading Time: 12 minutes Neste artigo, os autores apresentam uma breve descrição das técnicas de modelagem computacional de proteínas. Para uma melhor organização do artigo, o manuscrito foi dividido nas seguintes seções: introdução, métodos independentes de molde e métodos dependentes de molde. Revisão: BIOINFO – Revista Brasileira de Bioinformática. Edição #. . DOI: Neste artigo,…
Alinhamentos estruturais
Reading Time: 11 minutes Conheça os métodos de sobreposição de estruturas de macromoléculas

Alinhando estruturas por linha de comando com PyMOL
Reading Time: 3 minutes PyMOL é uma ferramenta de visualização de moléculas com uma grande quantidade de recursos em sua interface. Dentre esses recursos, se encontra o painel do terminal de linha de comandos. Por meio desse terminal é possível realizar inúmeras análises, como por exemplo, alinhamento de estruturas. Esse tipo de alinhamento pode ser…